
Введение
Саркома Юинга (лат. myeloma endotheliale) — злокачественная опухоль, формирующаяся в костях или мягких тканях. Саркома Юинга, как правило, поражает нижнюю часть длинных трубчатых костей, ребра, таз, лопатку, позвоночник и ключицу.
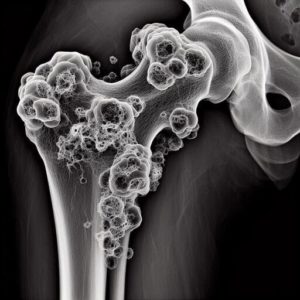
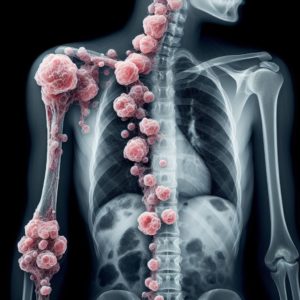
Определение саркомы Юинга
Саркома Юинга — это злокачественная опухоль костей, относящаяся к группе сарком мягких тканей. Она возникает из клеток надкостницы или костного мозга и характеризуется быстрым инвазивным ростом.
История открытия
Впервые саркома Юинга была описана американским патологоанатомом Джеймсом Юингом в 1921 году. Он выделил ее как отдельную нозологическую форму среди опухолей костей.
Распространенность
Саркома Юинга составляет около 10% всех злокачественных опухолей костей и около 1% сарком мягких тканей. Чаще болеют дети и подростки в возрасте от 5 до 20 лет. Ежегодная заболеваемость составляет 2-3 случая на 1 млн населения.
Виды саркомы Юинга
Различают несколько подтипов саркомы Юинга:
- Классическая саркома Юинга
- Периферическая примитивная нейроэктодермальная опухоль
- Askin опухоль (разновидность первой)
- Экстраоссальная саркома Юинга
Причины возникновения
Точные причины развития саркомы Юинга не установлены. Предполагается влияние генетических факторов, ионизирующего излучения, вирусных инфекций.
Факторы риска
- Возраст 10-20 лет
- Белая раса
- Наличие синдрома Дауна
- Лучевая терапия в анамнезе
- Химиотерапия алкилирующими агентами
Симптомы
- Боль в кости, усиливающаяся со временем
- Отек и припухлость над пораженным участком
- Ограничение подвижности сустава
- Перелом кости при минимальной нагрузке
- Усталость, слабость, снижение массы тела
- Лихорадка, озноб (при метастазах)
Диагностика
Для диагностики проводят:
- Обследование и пальпацию болезненного участка
- Рентгенографию
- КТ и МРТ для оценки распространенности
- Биопсию опухоли
- Анализ костного мозга
Патогенез
Генетические аспекты
Роль мутаций
Основную роль в развитии саркомы Юинга играют мутации в генах EWSR1 и FLI1. Они приводят к образованию химерного онкобелка, стимулирующего опухолевый рост.
Связь с опухолевым ростом
Химерный белок EWSR1-FLI1 активирует транскрипцию онкогенов, инициирует пролиферацию и подавляет апоптоз. Это запускает неконтролируемое деление клеток.
Влияние на клеточный цикл
Белок EWSR1-FLI1 нарушает регуляцию клеточного цикла, делая клетки нечувствительными к сигналам прекращения деления.
Молекулярные механизмы развития
Кроме химерного белка, важную роль играют мутации в генах супрессорах опухоли ТР53, Rb, и активация сигнальных путей PI3K/AKT/mTOR и RAS/RAF/MEK/ERK.
Классификация
По локализации опухоли
Чаще всего саркома Юинга поражает длинные трубчатые кости нижних конечностей и таза — бедренную, большеберцовую, малоберцовую, плечевую кости. Реже встречаются опухоли позвоночника, ребер, черепа.
Особенности каждого вида саркомы Юинга
Классическая форма чаще локализуется в диафизах длинных костей. Периферическая — в мягких тканях конечностей и грудной клетки. Askin опухоль поражает грудную клетку.
Прогностические факторы
Неблагоприятные факторы — метастазы, большой объем опухоли, поражение костей таза и позвоночника.
Клиническая картина
Стадии заболевания
Выделяют 4 стадии саркомы Юинга:
I — опухоль локализована в кости
II — есть метастазы в легких и костный мозг
III — метастазы в других органах
IV — диссеминированные метастазы
Симптомы на различных стадиях
На ранних стадиях преобладают боль, отек, ограничение подвижности. При прогрессировании — переломы, интоксикация, потеря веса, лихорадка. Метастазы в легких вызывают одышку и кашель.
Осложнения
Возможны патологические переломы, компрессия спинного мозга, развитие сепсиса.
Прогноз
Прогноз зависит от стадии, размера и локализации опухоли. 5-летняя выживаемость при I стадии составляет 70%, при IV стадии — менее 20%. Неблагоприятные факторы — метастазы, поражение осевого скелета, большой объем опухоли.
Диагностика
Инструментальные методы исследования
Рентгенологические признаки
На рентгенограмме определяется остеолитическое поражение кости с нечеткими контурами и периостальной реакцией.
КТ и МРТ
Позволяют оценить распространенность опухоли, наличие метастазов в легких и мягких тканях.
Биопсия
Проводится биопсия опухоли для морфологической верификации диагноза.
Лабораторная диагностика
Выявляет повышение СОЭ, ЛДГ, анемию, лейкоцитоз. Проводят ПЦР для обнаружения химерного гена.
Лечение
Хирургическое вмешательство
Радикальная резекция опухоли
Основной метод лечения — хирургическое удаление опухоли с чистыми краями.
Органосохраняющие операции
При возможности проводятся для сохранения конечности.
Показания к химиотерапии
Препараты платины, доксорубицин, циклофосфамид. Проводится до и после операции.
Роль лучевой терапии
Используется для повышения эффективности хирургического лечения.
Иммунотерапия
Применяются моноклональные антитела, ингибиторы контрольных точек. Усиливают противоопухолевый иммунитет.
Таргетированная терапия
Ингибиторы тирозинкиназ, mTOR и других молекулярных мишеней. Подавляют сигнальные пути опухолевого роста.
Прогноз и профилактика
Факторы, влияющие на выживаемость
Основные факторы прогноза — размер, стадия и локализация опухоли, радикальность оперативного лечения, метастазирование.
Рецидивы и метастазы
Рецидивы наблюдаются в 25-30% случаев, чаще в легких и костях. Проводится терапия второй линии.
Мониторинг после лечения
Регулярное обследование для раннего выявления прогрессирования — осмотр, рентген, КТ, анализы.
Меры по профилактике заболевания
Специфической профилактики не разработано. Важно своевременное обследование подозрительных симптомов.
Заключение
Современные тенденции в лечении саркомы Юинга
Активно развиваются органосохраняющие операции, таргетная терапия, иммунотерапия, новые схемы химиотерапии. Улучшается выживаемость пациентов.
Направления дальнейших исследований
Изучение молекулярно-генетических механизмов, поиск новых мишеней для терапии, разработка прогностических маркеров, создание вакцин.